
Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity
, , et al. (2008). Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. 105, 3041–3046.
Background
Mutations in the BRAF gene occur in a wide variety of human tumors. Specifically, the BRAF V600E missense mutation is the most frequent oncogenic protein kinase mutation known. Tumor phenotypes correlated with oncogenic BRAF mutations include increased severity of cancer and decreased response to chemotherapy. One therapeutic approach to treating BRAF mutant cancer involves direct inhibition of the oncogenic BRAF kinase activity. Thus, in BRAF V600E mutants, targeted inhibition of the BRAF V600E gene product may be an important avenue to treating such tumors, especially in traditional therapy-resistant tumor types. The development of highly specific and effective inhibitors of the BRAF V600E gene product would provide insight into the therapeutic relevance of this target in malignant tumors. In this paper, the authors did exactly such by discovering the BRAF V600E specific inhibitor PLX4720 and examining its efficacy in both cell and animal based models.
Scaffold- and Structure-Based Discovery of the Inhibitor
The paper started off with a discussion of the rational drug design process that led to the discovery of PLX4720. Scaffold and structure based discovery was utilized. In other words, protein kinase scaffolds were first identified for a selected library of 20,000 compounds. The compounds were applied at 200 µM to multiple solved, but structurally divergent protein kinases. The compound-kinase structures were then screened by co-crystallography to identify which compounds would best bind to the known kinase scaffolds. A tremendous amount of work went into such screening process as over 100 structures showing bound compound were solved in this manner. Once productive binding activity was discovered for a given compound, chemical analysis was then performed to modify the compound for further inhibitory binding to the protein kinase. Through such a characterization process, PLX4720 was discovered to selectively bind to the BRAF V600E gene product (See Figure 1 for the PLX4720 binding structure and other structures leading up to its discovery). In fact, the authors claim, based on IC50 analysis (that is, the concentration at which 50% of the kinase is inhibited) PLX4720 binds with greater than 10-fold selectivity for BRAF V600E than for the wild-type BRAF.

Modes of Inhibitor Interaction and Selectivity
After characterizing the discovery process, the authors move on to further describe the binding action between PLX4720 and BRAF V600E. Crystallographic analysis revealed that there may be multiple conformations to the structure’s bound state. Some of these conformations reflect the “active” state of PLX4720, that is, the state in which the compound inhibits BRAF V600E, while other conformations reflect an “inactive” state. Such conformations and their respective binding interactions are displayed in Figure 2.

Cellular Selectivity in Multiple Tumor Lines
PLX4720 was then tested for its selectivity for BRAF V600E in multiple mutant and wild type cell lines. Each cell line was incubated for 1 hour in various concentrations of PLX4720 treatment. Two outputs were then measured, the GI50 (50% of growth inhibition) of cellular proliferation and GI50 of P-ERK expression. A previously characterized MEK inhibitor, PD0325901, was used as a control to benchmark P-ERK expression levels. It was also shown that PLX4720 is even more selective for BRAF V600E in cell lines than in kinase alone reactions. In some cases, the selectivity was up to over 100 fold. In addition, the clear down-regulation of P-ERK indicated that PLX4720 critically affected BRAF-MEK-ERK pathway. Notably, the BRAF-MEK-ERK pathway is generally regulated by feedback mechanisms in wide-type tumor cell lines, that is, even with BRAF inhibition, P-ERK is not critically down-regulated. Interestingly, this is not the case in BRAF V600E mutants, as demonstrated by the critical down regulation of P-ERK. The authors speculate that the negative feedback loop is lost in BRAF V600E cells, resulting in a pronounced dependence on the BRAF-MEK-ERK pathway.
Melanoma cell biology
To further drive the point home, the authors then demonstrated the effect of PLX4720 inhibition on a larger panel of melanoma cell lines. P-ERK expression was examined by Western blotting, and representative blots were shown in Figure 3A. Of the cell lines tested, 1204Lu (a BRAF V600E line) and C8161 (a BRAF wild-type line) were selected for further study because these cell lines come from highly malignant tumors that were resistant to chemotherapy. After administering varying concentrations of PLX4720 to these cell lines, it was demonstrated that 1204Lu displayed lower cell viability, while C8161 was unaffected (Figure 3B). This indicated that PLX4720 may have potential clinical applications where traditional chemotherapy has failed. In Figures 3C and 3D, an Annexin V PI stain coupled with cell flow cytometry was utilized to characterize 1205Lu and C8161 when treated with different concentrations of PLX or with the same concentration of PLX at different time points. The Annexin stains for alive vs. apoptotic cells, while the propidium iodide (PI) stains for necrotic vs. non-necrotic apoptotic cells. The results indicate that under equal PLX4720 treatment conditions, more cells in the V600E line underwent apoptosis than cells in the wide-type line. Again, this is consistent with the authors’ claims that PLX4720 selectively inhibits BRAF V600E. In Figure 3E, a live dead assay using Calcein-AM and EtBr was applied under varying treatment concentrations of PLX4720. Once again, it is shown that the BRAF V600E mutant line was more critically affected by PLX4720 and had more dead cells than the wild-type line. In Figure 3F and 3G, synthetic skin was created using 1204Lu (BRAF V600e) and C8161(BRAF wild-type) lines. The synthetic skin were respectively treated with PLX and immunostained with DAPI (for DNA), S100 (indication of cell proliferation), PCNA (indication of cell proliferation), and TUNEL (for DNA fragmentation). In all stains indicating cell growth and viability, it is shown that there is weaker signal in the PLX4720 treated BRAF V600E mutant synthetic skin, while DNA fragmentation is shown to be increased under PLX4720 treatment. Meanwhile, all 5 stains appear to be relatively unchanged after PLX treatment in the BRAF wild-type synthetic skin. This again served to drive the nail in the coffin in saying that PLX4720 selectively inhibits BRAF V600E lines.

Animal Efficacy
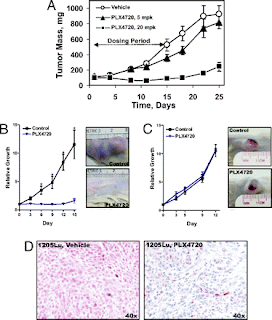
In addition to in vitro study, PLX4720 was further tested in mouse models for its efficacy to cause tumor regressions in vivo. A tumor xenograft of COLO205 cells (which are BRAF V600E mutant) was planted into nude mice. The xenograft mice then treated with either vehicle control, 5 mg/kg PLX4720, or 20 mg/kg PLX4720 by oral gavage daily on days 1-14 of the 25 day study. The results (Figure 4A) indicate that with increasing PLX4720 dosage, there was less tumor growth in the mice. In their style of driving the point home repeatedly, the authors then again performed xenografts using 1205Lu (BRAF V600E) and C8161 (BRAF wild-type) cell lines. Both were planted at two million cells into SCID mice, and the mice were treated with either vehicle control or 100 mg/kg PLX4720 by oral gavage twice daily. The results (Figure 4B and 4C) indicate that in the BRAF V600E tumors, PLX4720 caused significant tumor regression as there appeared to be little to no relative growth of the tumor, whereas in the BRAF wild-type tumor, PLX4720 appeared to have no effect. Furthermore, 1205Lu xenograft tumors were extracted from the mice, fixed in formaldehyde, paraffin embedded, and subject to immunostaining. Under immunostaining for P-ERK, the PLX4720 treated tumor extract indicated much lower P-ERK activity than the vehicle control. This indicates that PLX4720 not only selectively inhibits targets the BRAF V600E gene product in vitro (leading to tumor regression), but does so in vivo as well.
Conclusion
In brief, the authors make the argument that PLX4720 (a compound they had discovered) selectively inhibits the BRAF V600E gene product. Upon inhibition, cell viability is decreased and tumor regression appears to occur in both cell and mouse models. This may have large clinical impact for cancer treatment as BRAF V600E is a common mutation known to occur in many cancers. This study may also shed light on the mechanisms of cancer inhibition as the BRAF-MEK-ERK pathway is shown to be critically down-regulated in BRAF V600E lines under PLX4720 treatment.
Critique
The authors of this paper went to great lengths to drive their point home through a variety of methods pointing to the same conclusion. This is not uncalled for, as the authors make a rather convincing argument using the substantial amounts of evidence obtained from several different experimental techniques. Readers are left convinced that PLX4720 appears to be a potent BRAF V600E inhibitor that will have a large impact on clinical cancer treatment. Indeed, this paper has been widely cited, and PLX4720 has been referred 4 times in the New York Times to date.
One thing to be desired from this study is justification of the concentrations and time-points at which PLX4720 was administered. The authors appeared to choose arbitrary values best suited to demonstrate their point. Not much thought had been given to the clinical relevance of such values. Since the end goal of PLX4720 is to be developed as an avenue of clinical cancer treatment, choosing clinically relevant concentrations is critical. For example, in the mouse model studies, the concentration of PLX4720 varied from as low as 5 mg/kg to as high as 100 mg/kg. There can be a significant difference in toxicity between such values. In future studies (perhaps closer to clinical phase II stage), it would be appropriate to address approximate toxicity levels for the inhibitor.
The use of the BRAF-MEK-ERK pathway to indicate inhibitor selectivity was never fully justified. It is only empirically observed that BRAF V600E mutants tend to be more dependent on the BRAF-MEK-ERK pathway. The authors provide some speculation such as failure of the negative feedback mechanism, but no evidence is ever provided. Moreover, the use of COLO205 over other V600E lines in mouse xenografts was also not justified. However, to be fair, justification of the BRAF-MEK-ERK pathway dependence is truly difficult and probably beyond the scope of this paper. Also, in the case of mouse xenografts, the authors supplement their study by using both BRAF V600E mutant and BRAF wide-type xenografts.
![]()
5 comments:
It was really interesting to learn the techniques of discovering inhibitors to gene products. It was useful to learn all the factors they considered and testing conditions for validation; PLX4720 appears to be a promising remedy.
I was wondering if their animal efficacy tests were sufficient enough to reveal enough information about how PLX4720 works. They had to introduce cancerous cells specfically to nude/SCID mice, which will not have immunogenic reactions to xenografts; would performing tests on immune-deficient animals be adequate enough to show the big picture? I feel that it at least would not be enough to indicate proper dosages of PLX4720.
This is an interesting paper. I like how thorough it is in that it goes through so many techniques. I was wondering, in the animal experiments with nude mice, what effects does the drug have on the mice other than tumor regression? This kind of question would have implications for drug testing in other animals besides mice (ie, humans) and for the commercial use of the drug.
James -- Excellent point. I believe the authors only performed PLX4720 in vivo trials on a few mice as a very preliminary "proof of concept" to show that their drug has potential for in vivo studies, while their main point remains that they have discovered a really awesome BRAF inhibitor. I definitely agree that these mouse trials are not sufficient to indicate proper dosages. It will most likely to be up to other researchers and clinical scientists to perform further in vivo studies and clinical trials. If mouse trials are successful, believe proper dosing is determined on real patients during phase I clinical trials.
Mansi -- Great question, and I am very curious to that point as well. Generally, cancer drugs have a toxicity effect in addition to therapeutic potential. One major challenge is discovering inhibitors that are able to have high efficacy while still maintaining relatively low toxicity levels. Toxicity levels for a certain drug may also vary significantly from species to species, so I guess we won't know for sure until when and if this drug moves on to clinical trials.
The B-raf pathway is indeed an oncogenic pathway that is a downstream target of Ras. I think the BRAF-MEK-ERK pathway is an important Map kinase pathway that affects the transcription of early genes with transcription factors fos and jun. Perhaps this is the reason they chose to study this pathway? Additionally, I wonder how long it would take for tumor cells to mutate in order to become resistant to treatment by PLX4720. The authors state that the tumor cells in melanoma display oncogenic addiction to the B-raf pathway, which makes it vital for tumor survival. Overall, I thought it was a very interesting paper that shows a potential therapeutic application for PLX4720.
Post a Comment